Portugal implements EU medical device regulation

Portugal enforces Regulation (EU) 2017/745 on medical devices with Decree-Law no. 29/2024. This legislation covers rules for economic operators, healthcare institutions, and activities related to medical devices, ensuring compliance and public health protection.
On April 5, the Portuguese Official Gazette has published the Decree-Law no. 29/2024 (DL 29/2024) that enforces in Portugal Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices (Regulation 2017/745). DL 29/2024 establishes rules applicable to economic operators and to healthcare institutions that manufacture and use medical devices in their facilities, as well as the rules applicable to the use and traceability of devices, the designation and supervision of the activities of notified bodies, market surveillance and enforcement, and the sanctions applicable to non-compliance with these provisions.
1. National Authority
INFARMED, National Authority for Medicines and Health Products, I.P., is designated as the national competent authority for the purposes of Regulation 2017/745 and Decree-Law 29/2024. It is also responsible for monitoring and ensuring compliance with these regulations, as well as implementing public health protection measures.
2. Manufacturing activities
Manufacturers with domicile or registered office in Portugal who place custom-made devices on the market in their own name are required to notify INFARMED.
Among other obligations, DL 29/2024 stipulates that healthcare institutions that manufacture devices must:
- Notify INFARMED electronically, in the format made available for this purpose, regarding the devices they manufacture and use. This notification must be accompanied by supporting documents demonstrating compliance with a set of requirements established in DL 29/2024.
- Guarantee that their facilities, premises, equipment, software, utensils, raw materials, and other materials and substances used are suitable for the manufacturing process and the device manufactured, and that these are duly controlled to ensure the general safety requirements imposed by Regulation (EU) 2017/745.
- Report to INFARMED any relevant safety or performance issues related to the use of a device (e.g., any malfunction or deterioration of the characteristics or performance of a device).
- Establish proportionate financial coverage measures to ensure adequate compensation for potential liability arising from defective medical devices. This should be done in accordance with the producer liability regime for defective products (governed by Decree-Law No. 383/89 of 6 November).
- Appoint a technical manager with the specific competences necessary in the field of the devices in question.
In addition, regarding manufacture of devices by healthcare institutions it is not allowed to:
- Subcontract any manufacturing activity by the healthcare institution.
- Make the manufactured devices available to other healthcare institutions that are not part of the same legal entity.
- Place the device on the market in any form or affixing the CE marking to the device.
3. Use of medical devices
Within the scope of the rules on the acquisition, storage, and use of medical devices by healthcare institutions, it has been determined, among others, that it is now the responsibility of hospital pharmacy services, both public and private, to ensure the maintenance of conformity of medical devices, from their acquisition to their use.
Regarding the use of implantable medical devices, any entity, public or private, that uses implantable devices must:
- Provide the patient or consumer with the following information: (i) identification of the device (name, serial number, batch, unique device identifier, as well as the name, address, and website of the manufacturer), (ii) precautions and measures to be taken (external interference, medical examinations and environmental conditions), (iii) service life and follow-up (expected service life and necessary follow-up), as well as (iv) other information that ensures the safe use of the device.
- Record and keep the unique identification of the implantable devices used (an obligation that may come to cover other devices, by decision of the INFARMED board of directors).
It should be noted that for the purposes of acquisition, use, identification, and characterization of medical devices used by National Health Service (NHS) entities, it is necessary to present and register the device code, a matter to be regulated by Government order.
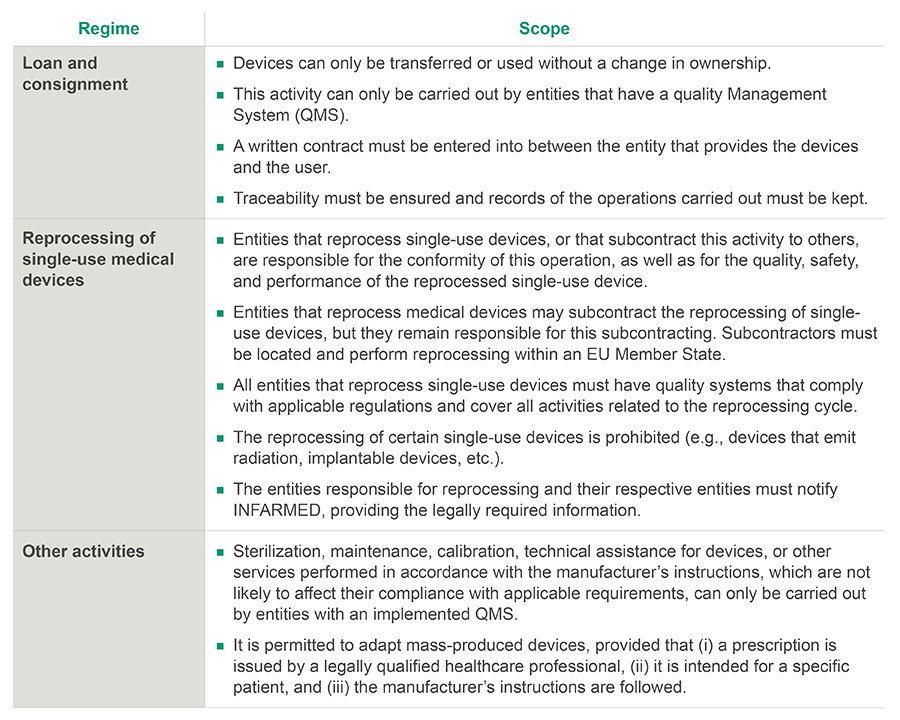
4. Other activities related to medical devices
DL 29/2024 regulates other activities within the medical device sector, namely:
5. Device distribution
- A notification system remains in place for those involved in supplying, possessing, storing or supplying devices intended for resale or use in medical services, healthcare units, pharmacies and other retail outlets, with the exception of direct supply to the public, under terms to be defined by INFARMED.
- Requirements for carrying out distribution activities include (i) a technical manager and (ii) adequate facilities and equipment with the capacity to ensure proper storage, preservation, and distribution of devices.
- Distributors are bound to comply with a wide range of obligations, which are aimed at, among others, ensuring the principles and standards of good distribution practices.
- The obligations imposed on distributors also apply to entities that carry out the first sale of devices to the public.
- National device manufacturers are exempt from notifying their activity for the distribution of devices they manufacture themselves, provided that certain requirements are met.
- The good distribution practices for medical devices (provided for in Order No. 256/2016 of September 28) are to be applied, with the appropriate adaptations, to healthcare institutions, regardless of their nature, including military hospitals.
6. General obligations
All operators that perform activities in the medical device sector (e.g., manufacturing, storage, marketing, display for sale, import, export, transport, making available, use in the provision of healthcare, or any form of device transaction) must:
- Ensure that the conditions of manufacture, storage, marketing, display, transport, use or transaction do not impair the conformity of the device with legal requirements.
- Not hold, possess, store, market, display, transport, use or transact devices in poor condition or that exceed their expiry date, with certain exceptions.
- Economic operators are obliged to cooperate with the competent authority, at its request, in actions aimed at eliminating or mitigating the risks associated with devices they manufacture, store, market, display, transport, make available, distribute or use in the provision of healthcare, or any form of device transaction.
7. Conformity assessment procedures
- The framework of conformity assessment procedures is governed by Regulation 2017/745. In contrast to its predecessor – the Decree-Law No. 145/2009 (DL 145/2009) –, which established the general framework for these procedures, DL 29/2024 does not address this area.
- However, INFARMED may, upon duly justified request, authorize the placing on the market or into service of specific devices even if they have not undergone conformity assessment procedures, provided that it is beneficial to public health or to the safety or health of patients.
8. National System for Surveillance of Medical Devices
- The National Medical Device Surveillance System remains in place. It is responsible for monitoring serious incidents that occur when people use medical devices. To do this, the System collects information on the safety of using these devices in humans, including their clinical, scientific, and technological evaluation.
- In cases of notification of serious incidents where the cause of the incident is related to or cannot be excluded from reprocessing, INFARMED may: (i) prohibit the use of the reprocessed device (ii) prohibit its reprocessing, and even (iii) determine the adoption of specific corrective measures to minimize the risk.
9. Transitional provision and entry into force
- DL 29/2024 came into force on April 6, 2024, and takes effect 90 days after its publication in the Diário da República, that is, on July 4, 2024.
- However, until EUDAMED (i.e., the European database on medical devices) is implemented and operational, some of the provisions of DL 145/2009 remain applicable to manufacturers, authorized representatives, and distributors. Specifically, with regard to wholesale distributors, the provisions of Order No. 256/2016 remain applicable, with the necessary adaptations.
- It should be noted that the legislator foresees the approval of two other distinct legislative acts:
- One relating to the advertising of medical devices. Thus, until the publication of this legislative act, the advertising regime provided for in DL 145/2009 remains in force.
- Another to ensure the implementation in Portugal of Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. In this regard, the provisions of Chapter XVII of DL 145/2009 remain in force until then.
- Without prejudice of the foregoing, Decree-Law 145/2009 is repealed.
Contacts


